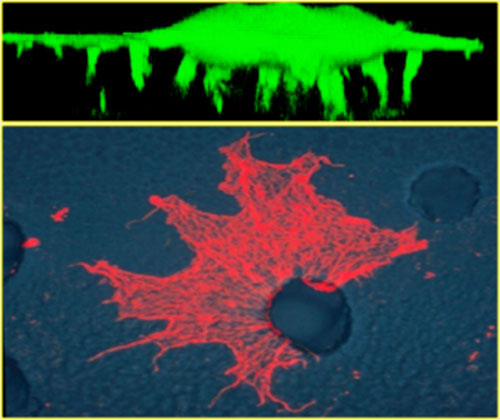
 Upper picture is a confocal image in the z plane of a green fluorescent cell protruding invadopodia membranes through the microporus filter (not visible). Lower picture is a cell protruding its invadopodia through a 3.0 um pore to the lower surface. Invadopodia are stained with rodamine-phalloidin to visualize the f-actin cytoskeleton.
Upper picture is a confocal image in the z plane of a green fluorescent cell protruding invadopodia membranes through the microporus filter (not visible). Lower picture is a cell protruding its invadopodia through a 3.0 um pore to the lower surface. Invadopodia are stained with rodamine-phalloidin to visualize the f-actin cytoskeleton.
Dr. Klemke's research program employs unique proteomic, phosphoproteomic, genomic and bioinformatics methodologies to identify novel phosphoprotein/kinase biomarkers (fingerprints) and map global kinase signaling networks that control cell polarization, immune cell homing, cell migration, proliferation, drug resistance, and cancer cell metastasis. To obtain the 'fingerprint' of metastatic cells, our lab has pioneered the development of important new subcellular membrane purification methods using microporous filters to uncover thousands of unique protein and gene signatures, which drive the metastatic behavior of cancer and immune cells. These molecular 'fingerprints' and their annotated signaling networks are being used as biomarkers to detect metastatic cells in the body and to evaluate the efficacy of novel anti-tumor and anti-metastatic agents using preclinical animal models of cancer.
Project 1: Invadopodia proteomics and metastatic signature identification: Pseudopodia and invadopodia are membrane projections made by invasive migrating cells that facilitate extracellular matrix degradation and migration through complex tissues in the body (Figure1). Cell migration during cancer metastasis requires morphological polarization characterized by formation of a leading pseudopodium (PD) at the front and a trailing rear at the back. This process is controlled by the spatial and temporal organization of complex protein signaling networks that partition to the front or back of the cell. Reversible protein phosphorylation on serine, threonine, or tyrosine amino acid residues is a major mechanism utilized by migratory cells to coordinate the localization, activation, stability, and assembly of macromolecular signaling complexes. However, aberrant regulation of protein localization and protein phosphorylation in cancer cells contributes to the processes of cell invasion, migration and metastasis. Using unique invadopodia/pseudopodia purification methods developed in the Klemke Lab, we have applied global proteome profiling in combination with quantitative phosphoproteomics and bioinformatics approaches for comparative analysis of the cell rear (cell body, CB) and invadopodia proteomes of invasive migrating cells. The spatial relationship of greater than 4000 proteins and hundreds of distinct sites of phosphorylation can be mapped revealing networks of signaling proteins that partition to the invadopodium and/or the cell body compartments. This powerful approach as revealed unique proteins and networks of phosphoprotein signatures that endow these cells with metastatic abilities. These protein signatures have important diagnostic and prognostic value, and are possible new therapeutic targets. Important new metastatic proteins have been identified and are being functionally characterized at the gene and protein level in the lab, including Lasp-1, PEAK1 tyrosine kinase, and eIF5A. Additional novel metastatic signatures and new phosphorylation sites have been identified and are ready for functional testing in cell-based and preclinical animal models of cancer. Computational and informatics analyses are also underway to understand how these metastatic signatures and kinase substrate interactions are integrated at the network level to control cell invasion and metastasis.
Project 2: PEAK1 and Cancer Progression. Using the invadopodia purification technology developed in our laboratory, we discovered pseudopodia-enriched atypical kinase one (PEAK1), a catalytically active tyrosine kinase that associates with the actin cytoskeleton and regulates cell migration and proliferation. PEAK1 is also an important scaffolding protein that couples integrin and growth factor receptor signaling to the cytoskeleton to control cell movement and proliferation. Figure one
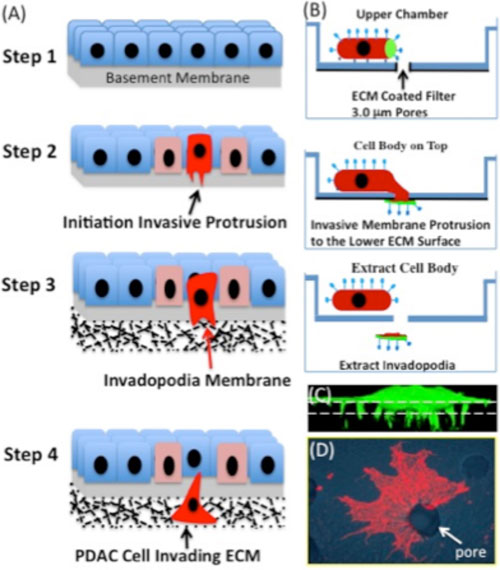 |
| Figure 1. (A) Schematic showing the steps of cancer cell invasion into the extracellular environment. Step1, normal healthy tissue architecture with an intact basement membrane. Step2, Genetic mutations drive cancer cell transformation, proliferation (pink cells), and loss of cell-cell contacts. Genetic mutations in cancer cells in association with changes in the tumor microenvironment drive formation of specialized actin-rich membrane protrusions, called invadopodia, which degrades the basement membrane (red cell). Step 3. Invadopodia transform into larger actin-rich membrane protrusions that degrade the surrounding extracellular matrix of the tissue. Step 4. These specialized membrane protrusions also provide propulsive forces and a steering mechanism to navigate through complex tissues in route to the circulation. (B) schematic illustration of invadopodia purification. Cells are allowed to attach to the surface of the upper chamber containing a 3.0 mm membrane filter. Extracellular matrix proteins coat the lower filter surface creating an adhesive gradient. The cells sense the matrix gradient and extend protrusions through the small openings to the lower surface in the direction of the ECM protein. The membrane protrusions can then be manually separated from the cell bodies, which remain on the upper membrane surface. The isolated cell bodies and invadopodia can then be profiled using proteomic methods. (C) Confocal image in the z plane of a GFP labeled cell protruding invadopodia through the microporous filter (dashed line). (D) An invadopodium with filapodia-like extensions protruding through a 3.0 mm pore (arrow) was fixed and stained with rodamine phalloidin to visualize the F-actin cytoskeleton. |
Several research projects are underway that use unique cell-based, genomic and proteomic methods to uncover gene and protein biomarkers of metastatic tumor cells:
 |
|
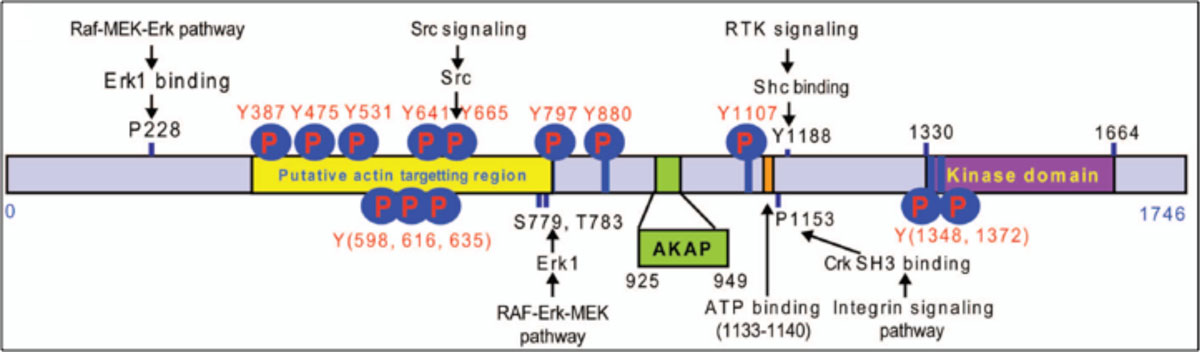
Figure 1. Pseudopodium-Enriched Atypical Kinase: PEAK1 (Sgk269)
Identities=379/690 (54%), Positives=464/690(67%)
RTK = receptor tyrosine kinase, P1153 = polyproline domain, Y665 = src phosphorylation and SH2 binding site. |
shows a schematic of how PEAK1's domain structures couple to the signal transduction machinery of cells. A substantial effort is now underway to understand how PEAK1 regulates cell migration, proliferation, and cancer progression.
Project 2a. Role of PEAK1 tyrosine kinase in pancreatic cancer formation and metastasis. Work pioneered in our lab has shown that PEAK1 is overexpressed in pancreatic ductal adenocarcinoma (PDAC) where it regulates tumor cell proliferation, metastasis and chemoresistance (Figure 1). Emerging work in the field indicates that this newly discovered kinase is a critical player in several human cancers including PDAC, colon, and breast cancer. To directly address the function of PEAK1 in normal physiology and cancer a conditional PEAK1 knockout animal has been generated in our lab using Cre-lox technology. Future projects will involve breeding the PEAK1 knockout mice to genetically engineered mouse models of PDAC to determine how PEAK1 contributes to cancer formation, progression and drug resistance. The normal physiological function of PEAK1 will also be investigated in these mice.
Project 2b. Dissecting the signal transduction role of PEAK1
The goal of these studies is to determine precisely how PEAK1 signaling (Figure 1) couples to the cell migration and proliferation machineries of normal and malignant cells. For these studies, we are using genetic engineering and site directed mutagenesis to perturb key signaling domains and phosphorylation sites in PEAK1. The mutant forms of PEAK1 are then tested for their ability to transmit various migration and proliferation signals within the cell's interior. Critical domains in PEAK1 that modulate src kinase signaling, adaptor protein binding, and kinase activity have been identified and functionally validated. PEAK1 signaling functions are also being investigated at the network level using PEAK1 knockout/knockin cells and powerful proteomic and phosphoproteomic profiling methods combined with computer-based informatics programs. Additional biochemical, mutagenesis, and proteomic works are in progress to identify specific PEAK1 binding partners and key effector pathways.
Project 2C. Role of PEAK1 in developmental and tumor-induced angiogenesis. New blood vessel formation (angiogenesis) is required for the sustained growth of primary and secondary metastatic tumor growth. These vessels provide life sustaining oxygen and nutrients that fuel new tumor growth. Therefore, inhibiting the process of tumor-induced angiogenesis is being intensely investigated as a possible means to inhibit tumor growth. Work in our lab has show n that PEAK1 tyrosine kinase plays a critical role in new vessel growth. In fact, Knockout of PEAK1 in zebrafish using TALEN technology (Transcription Activator-Like Effector Nucleases) or knockdown by specific PEAK1 morpholinos (MO) induced severe pericardial edema, blood-pooling defects, and disrupted the formation of intersegmental (ISV) and subintestinal vessels (SIV) (Figures 1 and 2). High-resolution intravital time-lapse imaging of the developing vasculature revealed that PEAK1 depletion inhibits both endothelial cell proliferation and migration of ISVs. Co-injection of suboptimal doses of PEAK1 and VEGF MO, which does not inhibit vessel development alone, induced a robust inhibition of ISV angiogenesis, suggesting PEAK1 has a synergistic role with VEGF. In further support of these findings, depletion of PEAK1 in HUVECs inhibits tube formation and vessel sprouting on fibrin-coated beads induced by VEGF-A. In addition, treatment of HUVECs with VEGF-A promoted phosphorylation of tyrosine 665 of PEAK1, a key Src kinase SH2 adaptor binding site. Finally, we found that PEAK1 expression is highly increased in the endothelium of neovessels of human tumor tissue samples compared to the quiescent endothelium of normal vessels suggesting that PEAK1 plays a role in tumor-induced angiogenesis. Together our findings indicate that PEAK1 is a novel tyrosine kinase necessary for angiogenesis. These findings also suggest that PEAK1 is a good therapeutic target to inhibit cancer, since it plays a dual role in tumor formation and tumor-induced angiogenesis. Signal transduction work is now underway to understand how VEGF receptor signaling activates PEAK1 to control endothelial cell proliferation and migration. We also have plans to investigate the role of PEAK1 in other vascular related diseases including retinopathy and wound healing.
Project 2D. Role of PEAK1 in cancer stem cell propagation and differentiation. Neoplastic tissues are believed to contain a subpopulation of stem-like cells with self-renewing capabilities (referred to cancer stem cells, CSCs). These cells are believed to be the metastatic culprits and the underlying cause of patient deaths due to relapse after chemotherapy and radiation. Therefore, new therapeutic approaches and strategies are sorely needed to target these highly resistant cells. Work in our lab has shown that PEAK1 is a new tyrosine kinase that controls the stem cell transcriptional machinery present in CSCs. Using a genetic PEAK1 knockout/Knockin approach, we have shown that PEAK1 signaling is the major driving component of a feed-forward transcriptional loop that controls the expression of the known stem cell regulators Oct-4 (Figure 1), c-myc, Soxs2, KLF4, Yap1, and Nanog. Perturbation of PEAK1 function inhibits the feed-forward loop, preventing tumor initiation and formation in vitro and in vivo. We are currently investigating in detail how extracellular cues in the tumor microenvironment integrate PEAK1 signaling with the CSC transcriptional program to drive tumor initiation and metastasis.
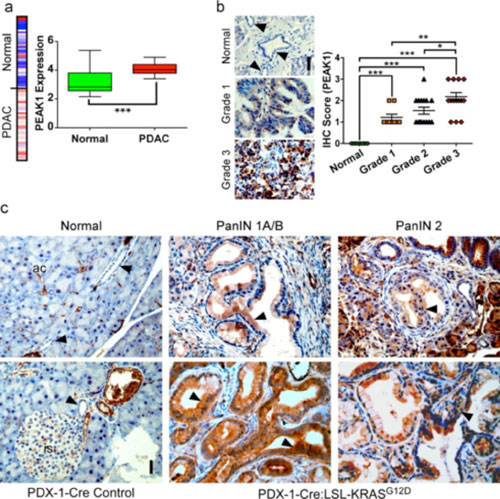 |
| Figure 1. PEAK1 is amplified in human PDAC and is an early biomarker for PanIN lesions and PDAC progression. (a) Heat map and Tukey plot of PEAK1 gene expression from a 39 patient microarray study analyzed using www.oncomine.org. (b) Immunohistochemistry (IHC) of PEAK1 in human PDAC or normal pancreatic duct tissue sections (left). Arrows indicate normal ductal epithelium. Scale bar equals 50 mm. Quantitative analysis IHC staining for 7 normal, 9 grade 1, 15 grade 2 and 16 grade 3 human samples using blind scoring on a scale of 0-3 (right). (c) IHC of PEAK1 in normal PDX-1-Cre control mice (ac = acinar cells, is = islet) and PanIN 1A/B, 2 and 3 from 3 or 6 month-old Pdx-1-Cre:LSL-KRasG12D mice. Arrows indicate normal ductal epithelium or PanIN lesions. Scale bar equals 50 mm. *, ** and *** represent p-values of <0.05, 0.01 and 0.001, respectively. |
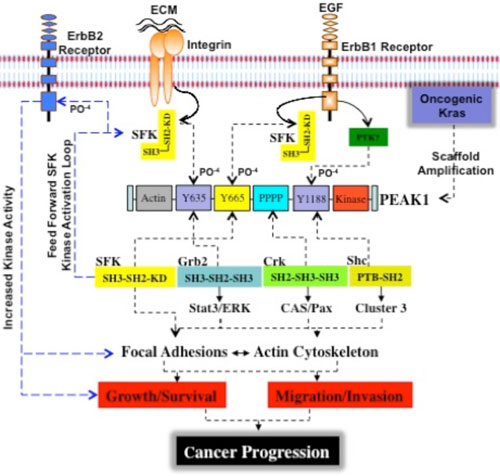 |
| Figure 1. Proposed model for PEAK1-mediated PDAC cancer progression and metastasis. In normal quiescent pancreatic cells, basal levels of PEAK1 and src kinase control homeostatic activities of integrin focal adhesions and the actin cytoskeleton to maintain epithelial shape and ductal architecture. In PDAC cases, oncogenic activation of Kras induces de novo ErbB2 tyrosine kinase expression and promotes the amplification of PEAK1 protein. This is a critical step as the aberrant increase in PEAK1 expression levels induce the kinase activated and open molecular conformation of Src, which is achieved through the binding of Src's SH2 domain to PEAK1's Y665 site. Interestingly, the PEAK1-Y665 site is not only a highly conserved Src SH2 binding consensus domain, but is also a highly conserved Src kinase consensus phosphorylation site. This serves as a mechanism to amplify and sustain Src kinase activity, which supports cell transformation and anchorage-independent growth of pancreatic duct cells. In this scheme, the activated PEAK1/Src kinase complex repeatedly interacts with numerous PEAK1 molecules driving intermolecular transphosphorylation of PEAK1-Y665 leading to more Src SH2 binding events and thus increased Src kinase activity. One of the major consequences of PEAK1-induced Src kinase amplification is binding to and phosphorylation of tyrosine 877 in the kinase domain of ErbB2. This has been reported to enhance its kinase activity, increase transforming potential, and further increase Src kinase activity. In this way, PEAK1/Src and ErbB2 co-associate, probably at the membrane-cytoskeleton interface, forming a triple kinase scaffold and robust kinase activation loop. PEAK1 is also a scaffold for several SH2/SH3 domain containing adaptor proteins including c-CrkII, Grb2, and Shc. The molecular assembly of a triple tyrosine kinase scaffold and adaptor signaling complex (PEAK1/Adaptors/ErbB2/Src) serves as an amplification mechanism to accelerate PEAK1 and Src activities toward downstream substrates and effectors including p130CAS (CAS), paxillin (Pax), Stat3, and ERK, which in turn couple to the proliferation and migration machineries of cells to drive cell growth, survival, and metastasis. ECM = extracellular matrix, SFK = src family kinase, SH2 = src homology two domain, SH3 = src homology three domain, EGF = epidermal growth factor. |
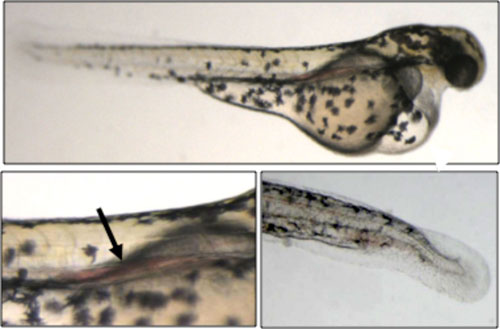 |
| Figure 1. Severe blood pooling defects caused by zPEAK1 knockdown. Embryos were injected with 3.2 ng zPEAK1 SP1 morpholino oligonucleotide at the one-cell-stage, and pictures were taken at 2 days post fertilization (dpf). Pericardial edema (upper panel) and blood accumulation in the anterior aorta (black arrow, left panel) are prominent. Blood accumulation in tail region can also be observed (bottom right panel). |
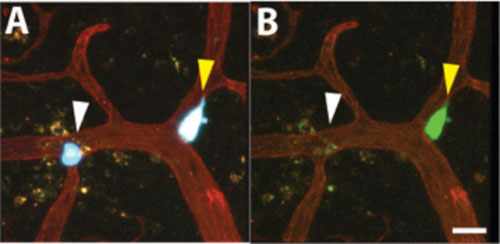 |
| Figure 1. Visualizing genetically encoded Oct-4 stem cell reporter activity in brain metastases. Cancer cells expression CFP (blue) and stably transduced with Oct-4-GFP (green) stem cell promoter activity reporter were injected into the chicken vasculature and allowed to colonize the brain. 3 days post injection the chicken vasculature was labeled with lectin-rhodamine (red) and the brain resected and imaged by multicolor confocal microscopy. Representative images show cancer cells expressing Oct-4-GFP reporter positive (yellow arrow) or negative (white arrow) cells that are attached to the chicken brain vasculature. (A) Red, green, and blue channels are shown. (B) Only red and green channels are shown. Note the absence of Oct-4 reporter expression in Oct-4 negative cell in B. Scale bar = 20 µm. |
Key References:
Wang, Y. and Klemke, R.L. Biochemical Purification of Pseudopodia from Migratory Cells. Methods in Molecular Biology. 370:55-66, 2007.
Wang, Y., Ding, S., Wang, W., Jacobs, J., Qian, W., Moore, R., Yang, F., Camp, D., Smith, R. and Klemke, R.L. Profiling signaling polarity in chemotactic cells. PNAS 104(20):8328-8333, 2007.
Wang, Y., Ding, S., Wang, W., Yang, F., Jacobs, J., Camp, D., Smith, D. and Klemke, R.L. Methods for Pseudopodia Purification and Proteomic Analysis. Sci.STKE 2007(400):14, 2007.
Wang, Y., Cao, H.S.T., Cantin, G.T., Lin, R., Wang, W., Kaushal, S., Kelber, J., Edgington, T.S., Hoffman, R.M., Bouvet, M., Yates, J.R. and Klemke, R.L. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression. PNAS, 107(24), 10920-5, 2010.
Kelber, J. and Klemke, R.L. PEAK1, a Novel Kinase Target in the Fight Against Cancer. Oncotarget, 1(3), 219-223, 2010.
Wang, Y., Yang, F., Huang, X., Wang, W., Jiang, X., Gritsenko, M.A., Zhao, R., Monore, M.E., Pertz, O.C., Purvine, S.O., Orton, D.J., Jacobs, J.M., Camp, D.G. 2nd, Smith, R.D. and Klemke, R.L. Spatial phosphoprotein profiling reveals a compartmentalized ERK switch governing neurite growth and retraction. J Biol Chem, 286(20), 18190-201, 2011.
Kelber, J., Reno, T., Kaushal, S., Metildi, C., Wright, T., Stoletov, K., Weems, J., Park, F., Mose, E., Wang, Y., Hoffman, R., Lowy, A., Bouvet, M., Klemke, R. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res.72, 2554-2564, 2012.
Bristow, J., Reno, T., Jo, M., Gonias, S., Klemke, R.. Dynamic phosphoryation of tyrosine-665 in pseudopodium-enriched atypical kinase 1 (PEAK1) is essential for the regulation of cell migration and focal adhesion turnover. J. Biol. Chem. 288, 123-131, 2013.
Kelber, J. and Klemke, R.. Proteomic and Biochemical Methods to Study the Cytoskeletome. Methods Mol Biol 1046; 203-18, 2013.
Choi, S., Kelber, J., Jiang, X., Strnadel, J., Fujimura, K., Pasillas, M., Coppinger, J., Klemke, R. Procedures for the biochemical enrichment and proteomic analysis of the cytoskeletome. Anal Biochem 1; 446:102-7, 2014.