Mechanistic insights provided by our experiments have led to the design and use of novel therapeutics that target the primary tumor, tumor-induced angiogenic vessels, and metastatic disease. In collaboration with leading medicinal and natural compound chemists at UCSD and in the corporate sector, our laboratory is characterizing new small molecule compounds, phytochemicals, and immune cell-based therapeutics (e.g NK, CAR-T Cells, etc.) that target metastatic tumor cells and their life sustaining blood supply. We are also investigating the mechanisms that lead to acquired and innate chemoresistance and have identified important signaling pathways and biomarkers that mediate this process. Several lead compounds, immune cell therapies, and natural treatments (nutraceuticals) have been identified that show promise, which are being developed for evaluation in early stage clinical trials.
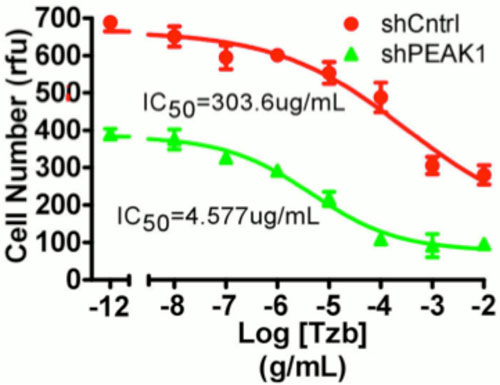
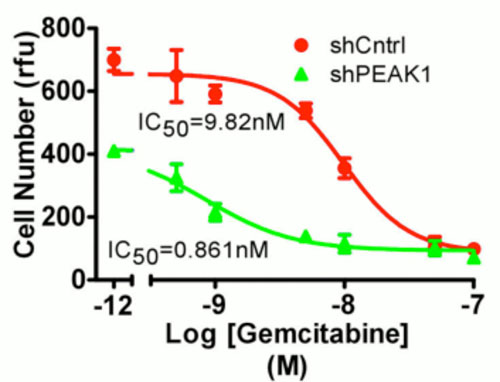 Genetic knockdown of PEAK1 tyrosine kinase (shPEAK1) increases sensitivity to the chemotherapeutic agent gemcitabine in gemcitabine resistant human pancreatic cancer cells. Control cells treated with random shRNA (shCntrl). IC50 = half maximal inhibitory concentration. RFU = relative fluorescent units.
Genetic knockdown of PEAK1 tyrosine kinase (shPEAK1) increases sensitivity to the chemotherapeutic agent gemcitabine in gemcitabine resistant human pancreatic cancer cells. Control cells treated with random shRNA (shCntrl). IC50 = half maximal inhibitory concentration. RFU = relative fluorescent units.
Several research projects are underway to identify lead compounds and investigate the mechanisms of drug action and chemoresistance:
Project 1. Therapeutic inhibition of the hypusine-eIF5A-PEAK1 switch, which regulates the pathogenesis of pancreatic ductal adenocarcinoma (PDAC). The overall goal of this project is to develop and commercialize a more effective treatment for pancreatic ductal adenocarcinoma (PDAC). Patients with PDAC have poor survival rates and limited treatment options. It is an extremely difficult-to-treat disease with a median survival of less than 1 year and a dismal 5-year survival rate of 3-5%. Currently, the chemotherapeutic drug gemcitabine is the first-line treatment for PDAC. However, gemcitabine does not significantly prolong survival and patients commonly develop chemoresistance and relapse. PDAC is characterized almost universally by mutations in KRas with frequent deregulations in embryonic signaling pathways such as Hedeghog (Hh) and Wnt--BioTheryx, we used a Drosophila model of Wnt signaling to identify ciclopirox olamine (CPX) (Figure 1), an FDA approved topical antifungal agent, as a novel modulator of this pathway. CPX also chelates iron and selectively inhibits non-heme iron enzymes such as deoxyhupusine hydroxylase (DOHH). DOHH catalyzes the final step in the unique posttranslational modification of the eukaryotic translation initiation factor 5A (eIF5A) with an unusual amino acid, hypusine (Figure 1). Given the fact that eIF5A is the only protein in nature that is modified by hypusine, and its selective upregulation in certain types of cancer, eIF5A represents an excellent therapeutic target for cancer. We have recently discovered that eIF5A is also deregulated in PDAC and controls PEAK1 (Pseudopodium Enriched Atypical Kinase 1) expression, which is an essential signaling molecule for PDAC growth and metastasis. (Figure 2) A direct correlation between gemcitabine resistance, eIF5A hypusination, and amplified PEAK1 expression was also identified in PDAC. As a known inhibitor of eIF5A hypusination and Wnt pathway, CPX showed in vitro antiproliferative activity in pancreatic cancer cell lines corroborated with decreased hypusinated eIF5A and PEAK1 expression. Mechanistically, CPX appears to be an ideal chemotherapeutic for PDAC, which is well supported by our in vitro studies. However, CPX is rapidly biotransformed by glucuronidation making the development of a systemic CPX (oral or injectable) therapy for PDAC difficult. Therefore, our current focus with BioTheryx is on the development of a CPX prodrug that has significantly better in vivo bioavailability in order to overcome some of the issues with delivery of the drug to a solid tumor such as PDAC. Work is underway to investigate the in vivo pharmacokinetics, safety, and efficacy of the CPX prodrug, BTX702, in animal models of PDAC.
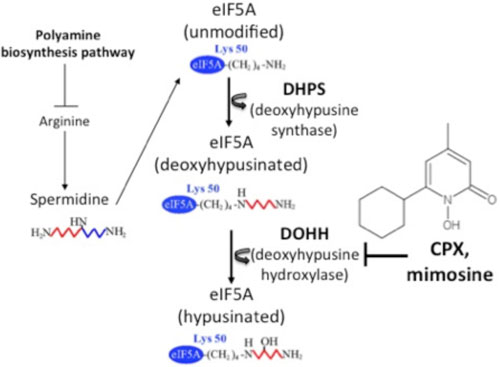 |
| Figure 1. Hypusine modification of eIF5A. The polyamine biosynthesis pathway mediates the generation of spermidine from arginine, a dietary precursor of polyamines. Hypusination involves two sequential enzymatic reactions. DHPS (deoxyhypusine synthase) transfers the 4-aminobutyl moiety from spermidine to the ε-amino group of a specific lysine residue (Lys-50 in the human protein) in eIF5A to generate eIF5A intermediate, which is subsequently hydroxylated by DOHH (deoxyhypusine hydroxylase) to form the mature, hypusinated form of eIF5A. CPX (ciclopirox oloamine), an iron-chelating hydroxypyridinone that is approved by the FDA as an anti-fungal agent, potently inhibits eIF5A hypusination by suppressing DOHH activity. |
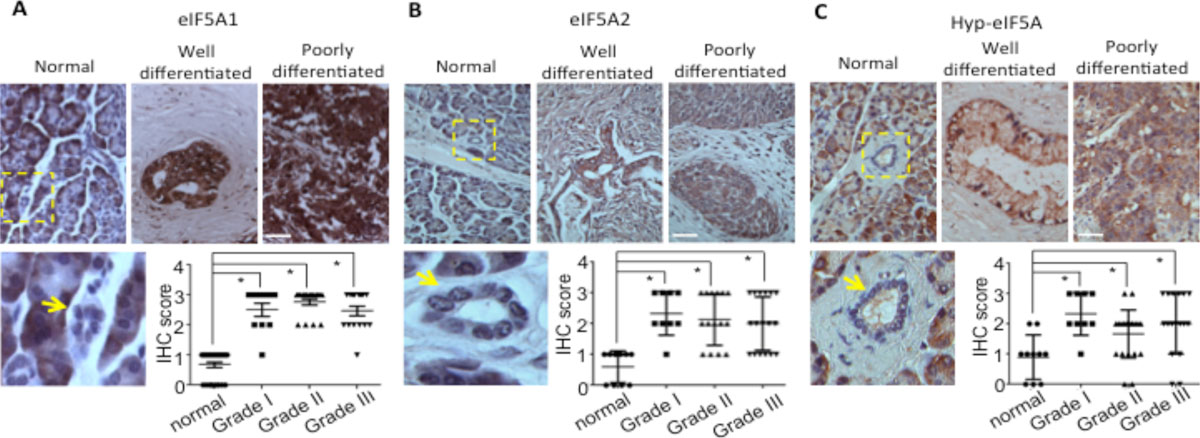 |
| Figure 2. Immunohistochemical staining of eIF5A1 (A), eIF5A2 (B) and Hyp-eIF5A1 (C) in normal human pancreatic tissues and PDAC tissues of varying tumor grades. A-C, Graphs show quantitative analyses of blind scoring on a 0-3 scale. A-C, arrows indicate normal pancreatic ducts or tumor tissues and boxed regions show normal ducts with corresponding high magnification images. Bars = 100 mm. * represents P values of < 0.05 as determined by Student's t-test. |
Project 2. Deciphering gemcitabine chemoresistance mechanisms in PDAC. Many cancers relapse due to the generation of chemotherapy resistance (chemoresistance) rendering first-line treatment drugs ineffective. In fact, the low response rates and poor clinical outcomes for many patients can be attributed to innate or acquired chemoresistance. However, the mechanisms and related biological pathways that contribute to chemoresistance are poorly understood. While Gemcitabine is the first-line chemotherapy for PDAC, approximately 40% of patients display resistance or acquire resistance during the course of treatment. Work in our lab has shown that exposure of gemcitabine resistance human PDAC cells to this drug induces hypusine activation of the eIF5A-PEAK1 signaling axis as described above in project 1. Importantly, genetic or pharmacological inhibition (CPX/BTX702) of eIF5A-PEAK1 signaling in these cells re-sensitizes them to gemcitabine-mediated killing (Figure 1). These findings suggest that therapeutics like CPX/BTX702 that target the eIF5A-PEAK1 signaling axis may be effective treatments for PDAC, when administered alone or in combination with gemcitabine. Also, our findings suggest that screening PDAC patients for eIF5A-PEAK1 response and activity levels may be an effective means to identify and stratify gemcitabine sensitive and resistance patients. Work is now underway to decipher the molecular signaling mechanisms and tumor cell heterogeneity that drive gemcitabine-mediated eIF5A-PEAK1 activation in PDAC and other cancers. We are also testing whether gemcitabine and BTX702, when administered as a combination therapy, can synergistically inhibit PDAC tumor development using preclinical animal models of PDAC.
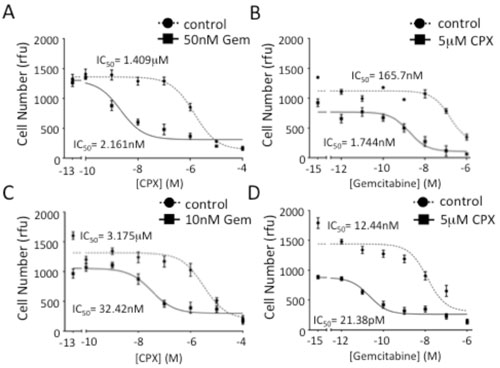 |
| Figure 1. Inhibiting eIF5A function in PDAC cells increases gemcitabine sensitivity. A, Analyses of viable cell number after 7 days of growth for PANC1 cells treated with increasing doses of CPX in the presence of 50 nM gemcitabine. B, Analyses of viable cell number after 7 days of growth for PANC1 cells treated with increasing doses of gemcitabine in the presence of 5 M CPX. C, Analyses of viable cell number after 7 days of growth for FG cells treated with increasing doses of CPX in the presence of 10 nM gemcitabine. D, Analyses of viable cell number for FG cells performed as in B. IC50 values represent the concentration of a drug that is required for 50% inhibition of cell proliferation as compared to vehicle-treated cells. Rfu=relative fluorescence unit. |
Project 3. Deciphering trastuzumab chemoresistant in PDAC. Trastuzumab (Herceptin, Genentech Inc.) acts to cause internalization, degradation, and inactivation of the ErbB2 receptor and is currently being evaluated in combination with gemcitabine, which is the standard chemotherapy given to patients with PDAC. However, studies to date suggest that trastuzumab offers little benefit alone or in combination with gemcitabine. These data, combined with the fact that PEAK1 is upregulated in trastuzumab-resistant PDAC cell lines and patients with amplified ErbB2 levels, led us to investigate whether the ineffectiveness of trastuzumab-based therapies could be due to a PEAK1/Src compensation mechanism following ErbB2 suppression. Trastuzumab treatment of PANC1 cells (a trastuzumab-resistant line) reduced ErbB2 expression significantly and increased PEAK1 expression, Src activity, and robust Y877 phosphorylation of the residual ErbB2 protein, which is a measure of kinase activity. PEAK1 knockdown abrogated this effect and greatly sensitized these cells to trastuzumab. While overexpression of PEAK1 in HPNE-KRas cells did not significantly change their response to trastuzumab, PEAK1 knockdown enabled trastuzumab to elicit growth-inhibitory effects under anchorage-dependent conditions (Figure 1). Rescuing PEAK1 expression in the HPNE-KRas cells reversed this effect, causing these cells to regain their trastuzumab-resistant phenotype. Furthermore, PEAK1 knockdown reduced the IC50 values of trastuzumab and a commercially available ErbB2-targeting antibody by approximately 100- and 10-fold, respectively, whereas PEAK1 overexpression in FG cells made them nonresponsive to trastuzumab in vitro. These findings support a mechanism by which elevated PEAK1/Src signaling compensates for loss of ErbB2 function in PDAC and, therefore, contributes to resistance of ErbB2-targeted therapies. Together our findings point to PEAK1 activation as a major mechanism that contributes to trastuzumab as well as gemcitabine resistance (project 2 above). In this regard, pharmacological studies are now underway to determine if blocking eIF5A-PEAK1 activities with CPX/BTX702 can reverse trastuzumab resistant in cell-based assays as well as preclinical animal models of PDAC. We are also concomitantly investigating the underlying molecular mechanisms responsible for eIF5A/PEAK1/Src activation using trastuzumab resistant and sensitive cell model systems.
 |
| Figure 1. Blocking VEGFR kinase activity, inhibits tumor-induced angiogenesis in zebrafish. Human cancer cells (red) secreting VEGF were allowed to form tumors for four days post injection (dpi) and induce a vascular supply (green) in the peritoneal cavity of Tg(fli1:egf) transgenic zebrafish. The same tumor was imaged before (right panel) and after treatment (24hrs, 5dpi) (left panel) with the VEGF kinase inhibitor SU5416. Note the significant loss of vessels and the dramatic decrease in vessel diameter. |
Project 4: Identification and characterization of novel anti-cancer and anti-angiogenesis inhibitors using preclinical animals models. The zebrafish model of angiogenesis has emerged as a powerful, simple, and cost effective model to uncover new small molecules and natural compounds that perturb developmental angiogenesis (Figure1). The molecular and genetic mechanisms that drive angiogenesis in this animal have been well described and recapitulate many of the same biological processes that drive VEGF-induced angiogenesis in many human cancers. Also, heart rate and drug toxicity can be easily monitored in this model organism. We are working closely with academic and corporate researchers to screen synthetic small molecule and natural compound libraries. Several promising new inhibitors have been identified and are being evaluated in our preclinical animal models of cancer and human clinical trials with great success. For more information on drug development research in the Klemke lab, please contact Dr. Klemke (rklemke@ucsd.edu).
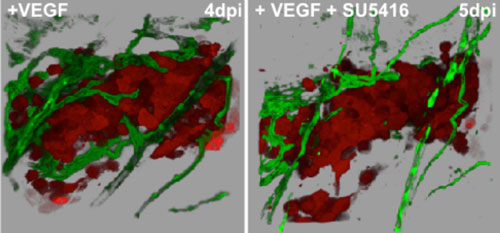 |
| Figure 1. Blocking VEGFR kinase activity, inhibits tumor-induced angiogenesis in zebrafish. Human cancer cells (red) secreting VEGF were allowed to form tumors for four days post injection (dpi) and induce a vascular supply (green) in the peritoneal cavity of Tg(fli1:egf) transgenic zebrafish. The same tumor was imaged before (right panel) and after treatment (24hrs, 5dpi) (left panel) with the VEGF kinase inhibitor SU5416. Note the significant loss of vessels and the dramatic decrease in vessel diameter. |
Key References:
Kelber, J., Reno, T., Kaushal, S., Metildi, C., Wright, T., Stoletov, K., Weems, J., Park, F., Mose, E., Wang, Y., Hoffman, R., Lowy, A., Bouvet, M., Klemke, R. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res.72, 2554-2564, 2012.
Stoletov, K., Montel, V., Lester, R., Gonias, S. and Klemke, R.L. High-Resolution Imaging of the Dynamic Tumor Cell-Vascular Interface in Transparent Zebrafish. PNAS. 104(44):17406-17411, 2007.
Wrasidlo,W., Mielgo, A.,Torres, V.A., Barbero, S., Stoletov, K., Suyama, T.L., Klemke,R.L., Gerwick, W.H., Carson, D.A. and Stupack, D.G. The marine lipopeptide somocystinamide A triggers apoptosis via caspase 8. PNAS. 105(7):2313-18, 2008.
Stoletov, K., Klemke, R.L. Catch of the day: zebrafish as a human cancer model. Oncogene. 27(33):4509-4520, 2008.
Jin, H., Garmy-Susini, B., Avraamides,C., Stoletov,K., Klemke,R. & Varner,J. A PKA-Csk-pp60Src signaling pathway regulates the switch between endothelial cell invasion and cell-cell adhesion during vascular sprouting. Blood, 116(25), 5773-83, 2010.
Murphy, E., Shields, D., Stoletov, K., Dneprovskaia, E., McElroy, M., Lindquist, J., Acevedo, L., Anand, S., Majeti, B., Tsigelny, I., Saldanha, A., Walsh, B., Hoffman, R., Bouvet, M., Klemke, R.L., Vogt, P., Arnold, L., Wrasidlo, W. and Cheresh, D. Disruption of angiogenesis and tumor growth with an orally active drug that stabilizes the inactive state of PDGFRβ/B-RAF. PNAS. 107(9):4299-4304, 2010.
Stoletov, K., Kato, H., Zardouzian, E., Kelber, J., Yang, J., Shattil, S. and Klemke, R. Visualizing Extravasation Dynamics of Metastatic Tumor Cells. J Cell Sci, 123(Pt13), 2332-41, 2010.
Stoletov, K., Strnadel, J., Zardouzian, E., Momiyama, M., Park, F., Kelber, J., Pizzo, D., Hoffman, R., Vandenberg, S., Klemke, R. Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 126, 904-913, 2013.